Hlízotvorné Solanum, které představuje kulturní brambor a jeho plané příbuzné druhy, je velice rozmanitá skupina (HAWKES, 1990). Většina těchto planých druhů obsahuje celou řadu neznámých systémů odolností vůči chorobám a škůdcům, mrazuvzdornosti, suchovzdornosti aj.
, které se v kulturních formách bramboru vůbec nevyskytují. V jihoamerické a v mexické oblasti existuje řada druhů s odolností k virózám bramboru, popř. s odolností vůči přenašečům těchto viróz, jsou známy druhy, které například nejsou napadány mandelinkou bramborovou. Výskyt genů zodpovědných za tyto vlastnosti je podmíněn především areálem výskytu jednotlivých druhů a podmínkami, jež zde panují.
Genová banka VÚB v Havlíčkově Brodě uchovává kromě odrůd bramboru a různých hybridních materiálů i velmi cenné genové zdroje planých druhů rodu Solanum. V tomto souboru je 22 druhů, které hlízy tvoří, a jeden druh, který hlízy netvoří. Druhy jsou udržovány ve více vzorcích a byly získány ze světových kolekcí genetických zdrojů bramboru (HORÁČKOVÁ a DOMKÁŘOVÁ, 2003). Popis morfologických znaků, hodnocení růstové energie, vegetační doby, zdravotního stavu, výnosu a vybraných hospodářských vlastností je prováděn podle „Klasifikátoru pro genus Solanum L.“ (Vidner et al., 1987).
Brambor je jednou z nejdůležitějších potravinářských plodin na světě. Zlepšení jeho vlastností proto může mít významný dopad. Čtení genomové sekvenence bramboru je však velmi složité, protože běžné brambory jsou tetraploidní, tj. jsou složeny ze čtyř sad chromozomů, což mimo jiné ztěžuje správné poskládání referenčního genomu. V současnosti je již také dostupná referenční sekvence v sestavení po chromozomech ve verzi v4.04 (Hardigan et al., 2016). Velmi přesná genomová sekvence umožňuje rychlejší a cílenější šlechtění, protože v DNA je snazší najít, která křížení s jinými odrůdami by mohla být zajímavá a kde by výměna genetického materiálu mezi „otcem“ a „matkou“ měla v ideálním případě probíhat. To znamená, že šlechtitelé vědí v rané fázi, zda brambor má požadované vlastnosti, jako je např. odolnost vůči specifickým chorobám.
V genetice je hojně využívaným nástrojem genome-wide association study, tj. celogenomová asociační studie (studie GWA nebo GWAS), srovnávací studie genetických variant u různých jedinců, která určuje úroveň spojení mezi genetickou variantou a vlastností. GWAS se obvykle zaměřují na asociace mezi jednonukleotidovými polymorfismy (SNP) a fenotypovými vlastnostmi, jako jsou významná geneticky podmíněná lidská onemocnění, ale mohou být stejně aplikovány na jakékoli jiné genetické varianty jakýchkoli jiných organismů a jejich fenotypové znaky.
Standardní znázornění výsledku GWAS je prováděno skrze Manhattan graf. Každá tečka grafu představuje SNP, přičemž osa X ukazuje polohu v genomu a osa Y ukazuje úroveň asociace.
Studie GWA zkoumají celý genom, na rozdíl od metod, které specificky testují malý počet předem určených genetických oblastí. Studie GWA identifikují SNP a další varianty v DNA spojené s onemocněním nebo jiným vybraným znakem, nemohou však samy definitivně určit, které geny jsou příčinné.
Materiál a metody
Z genové banky bramboru byl vybrán soubor fenotypově charakterizovaných genotypů bramboru s rozdílnými hodnotami pro barvu dužniny, ranost, obsah škrobu a rezistence k vybraným biotickým a abiotickým činitelům. Tyto rostliny byly průběžně pasážovány a vedeny jak v podmínkách in vitro tak i in vivo. Tento soubor byl doplněn fenotypově částečně charakterizovanými šlechtitelskými materiály, u kterých jsou tato data v průběhu řešení projektu doplňována a současně srovnávána s genotypovými daty.
DNA byla extrahována z 376 genotypů bramboru na pracovišti ÚEB AV ČR Olomouc, v.v.i., kde byl čerstvý materiál nejprve lyofilizován, a poté homogenizován pomocí skleněných kuliček. Pro vlastní izolaci byl použit NucleoSpin Plant II kit (Macherey-Nagel). Byla zkontrolována kvalita DNA a poté každý vzorek naředěn na požadovanou koncentraci. Izolovaná DNA byla zaslána do Diversity Arrays Technology Pty Ltd, Canberra, Austrálie, k vlastní analýze genotypu pomocí DArTSeq analýzy. Výsledkem DArTseq analýzy jsou celogenomová genotypová data SNP polymorfismů pro 372 vzorků. Genotypová data byla dále filtrována na chybějící informace, frekvenci minoritní alely a mapována k aktuální verzi genomu bramboru.
Celogenomová asociační analýza byla provedena pro 8 vybraných fenotypových znaků (odolnost k rakovině bramboru, odolnost k háďátku, ranost, obsah škrobu, odolnost k Viru Y bramboru ( PVY), odolnost k Viru svinutky bramboru (PLRV), výnos a suchovzdornost) podle dostupných dat hodnocení. Asociační analýza byla provedena pomocí programu GAPIT implementovaného v jazyce R. Pro výpočet byl použit statistický výpočetní modelBlink (HUANG et al. 2019) s korekcí na genetickou strukturu populace, genetickou příbuznost a další matoucí efekty. Spolehlivost výsledků asociace byla posouzena pomocí kvantil-kvantil Q-Q grafu při porovnání očekávaných výsledků proti získaným. Výsledky asociace polymorfismů a jejich fyzická pozice byly vyneseny do Manhattan grafů.
Výsledky a diskuse
Výsledky asociačních analýz celogenomových genotypů bramboru prokázaly vazbu mezi fenotypovým projevem a sekvenční variabilitou genotypů brambor. Znalost markerů asociovaných s požadovaným znakem umožňuje sledovat prostupnost genetické informace z rodičů na potomky napříč generacemi. U barvy dužniny můžeme například využít znalost asociovaného markeru k cílenému přikřížení znaku barvy pomocí zpětného křížení, tak abychom co nejlépe zachovali genetické pozadí obohacovaného genotypu. U jiných znaků může znalost asociovaného markeru umožnit šlechtitelům dříve a snáze rozpoznat tuto zájmovou vlastnost již v raných fázích vývoje potomků. Přiložené grafy reprezentují výsledky výše uvedené asociace jednotlivých vazeb mezi vybranými fenotypovými projevy a genotypovým profilem. Podrobný popis je uveden v naší předešlé práci (Ptáček et al., 2020), kde je popsán i význam jednotlivých grafů.
Přiložené grafy (Obrázek 1) prezentují výše uvedené asociace, Q-Q graf výsledků asociace potvrzuje statistickou průkaznost výsledků. Výsledky asociace k odolnosti k háďátku nejsou na uvedeném obrázku prezentovány. Pro posouzení asociace byl použit statistický model Blink. Model Blink - Bayesian-information and Linkage-disequilibrium Iteratively Nested Keyway (HUANG et al. 2019) využívá nový postup opakovaného hodnocení asociace, který vybírá v hodnoceném lokusu v genomu to SNP, které nejlépe vyjadřuje změny v hodnotách hodnoceného znaku. Dále byly hodnoty asociace jednotlivých SNP korigovány podle genetické příbuznosti položek mezi položkami a podle genetické struktury, která byla již dříve rozpoznána v hodnocené kolekci brambor. Polymorfismy SNP označená touto metodou za nejvíce asociované v jednotlivých lokusech jsou obzvlášť vhodné pro jejich použití v markery asistované selekci, neboť jsou očištěny od vlivu ostatních blízkých SNP. Paralelně s přicházejícími výsledky asociačních studií byl zahájen převod vybraných SNP u některých znaků na PCR markery, které budou použity ve šlechtění.
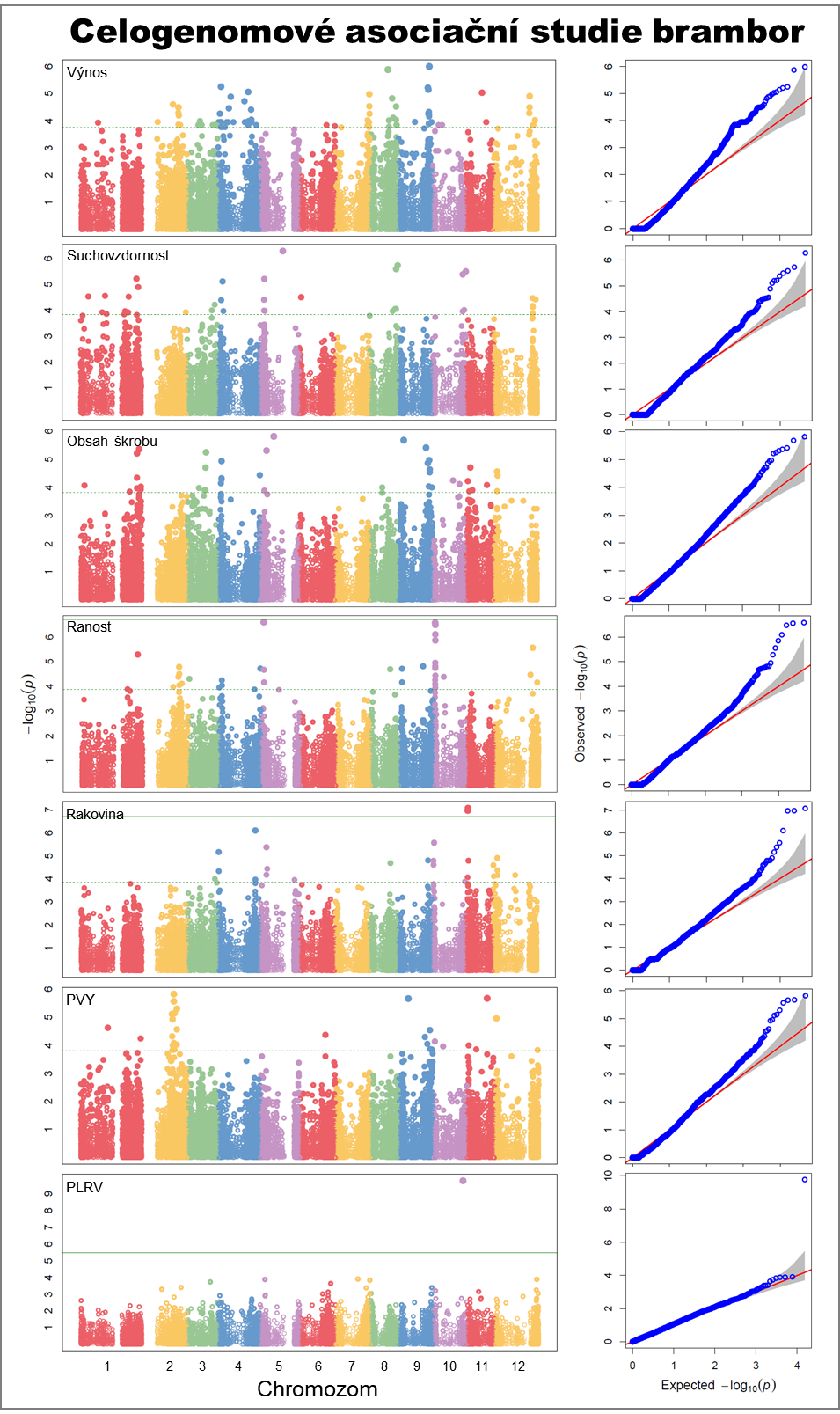
- Obrázek 1 V levé části obrázku jsou tzv. Manhattan grafy, které graficky znázorňují genotypovou asociaci k sledovanému znaku plodiny. Každý bod představuje jednonukleotidovou záměnu v řetězci DNA tzv. SNP. Úroveň asociace SNP je určena jeho vertikální pozicí v grafu. V horizontálním směru je znázorněna pozice SNP v genomu brambor. Barevně jsou rozlišeny jednotlivé chromozomy genomu lilku bramboru. Úroveň asociace je vyjádřena pomocí záporného dekadického logaritmu P-hodnoty statistického testu vypočtenou metodou BLINK. Zelená čára je hranice statistické významnosti P-hodnoty upravené na mnohonásobné testování na úrovni FDR = 0,05 (přerušovaná) nebo FDR = 0,01 (plná). V pravé části grafu jsou příslušné Q-Q grafy znázorňující distrubuci hodnot úrovně asociace pozorovaných a očekávaných, červená čára kopíruje teoretickou hladinu očekávaných hodnot, velká odchylka vyšších pozorovaných hodnot od hodnot očekávaných značí vysokou naměřenou míru asociace mezi genetickým polymorfismem a sledovaným znakem.